



To My Quyen,
Nguyen Khanh Thuan, Nguyen Phuc Khanh, Nguyen Thanh Lam*
1. Introduction
Edema disease (ED) is a systemic disease of weaned piglets caused by host-adapted strains of Escherichia coli (E. coli) that produce a variant of Shiga toxin 2, Stx2e. This toxin is closely related to Stx2, produced by human isolates of E. coli capable of causing hemolytic uremic syndrome (Paton and Paton, 1998). Clinical signs of edema disease include swollen eyelids, neurological symptoms (such as ataxia and stumbling), recumbency, and/or sudden death. These E. coli strains colonize the ileum via host-specific F18 fimbriae. Many edema disease isolates also produce heat-stable toxins that cause diarrhea. Historically, edema disease has been caused by E. coli strains belonging to serogroups O138, O139, and O141. Evidence suggests that these E. coli strains can be transmitted from both the sow and the environment to the piglets (Helgerson et al., 2006).
2. Aetiology
2.1 Bacterial characteristics
Edema disease is caused by Stx2e produced by a specific virotype of E. coli (STEC) (Figure 1). A virotype is determined by a particular combination of virulence genes. Important virulence factors encoded by STEC are the fimbrial adhesin F18 which is needed for attachment and colonisation of the gut and Shiga toxin (Stx2e) causing systemic disease. A different pathogenic E. coli is enterotoxigenic E. coli (ETEC). The ETEC carry the fimbriae F4 and F18, which again allow adhesion and colonisation of the gut, which results in production of enterotoxins including heat stable toxin a (STa), heat stable toxin b (STb) and heat labile toxin (LT) which cause post-weaning diarrhoea. The pigs need to have specific porcine F18 receptors (F18R) on their enterocytes for the STEC to bind (Coddens et al., 2007).

2.2 Classification
Basic classification
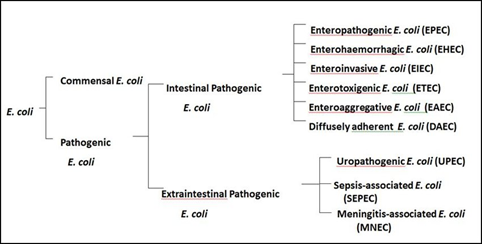
Intestinal pathogenic E. coli consist of several pathotypes: enteropathogenic, enterohaemorrhagic, enteroinvasive, enterotoxigenic, enteroaggregative and diffusely adherent all of which cause diarrhoea in both humans (all pathotypes) and animals, mainly enterotoxigenic and enteropathogenic (Figure 2) (Bélanger et al., 2011)
Serotype classification
Serotypes are based on the specific combination of the somatic O-antigen (O) and either flagellar (H), fimbriae (F) or capsular surface (K) antigens (Kaper et al., 2004). Whilst serotyping is established using specific combinations of at least 2 different O, H, K or F antigens, serogrouping is centred on O-antigen. Serotyping however, has many limitations; chief among these is that E. coli is the most difficult and time consuming genus of the Enterobacteriaceae family to serotype. This is because uncharacterised strains often require identification of all O, F and K serogroups. Due to the vast number of cross-absorbed antisera (167 O-antigen, 56 F-antigen and 74 K-antigens) required to type E. coli, full serotyping is only attempted by international reference laboratories. The genes involved in the biosynthesis of O-antigens are assembled together in a region called the rfb gene cluster between two housekeeping genes galF and gnd. As rfb clusters typically contain one unique gene linked to a serogroup, serogroup specific PCRs can be developed to replace conventional serological serogrouping (Reeves et al., 1996; Wang et al., 2001).
3. Epidemiology
3.1 Epidemiologic aspects
Edema disease is mainly seen in pigs 7 to 10 days after weaning. The disease usually occurs as sporadic cases or small outbreaks in pigs of a given age. Morbidity is relatively low and mortality is high. A morbidity rate of 16% (range: 10% ‒ 35%) and a case mortality rate of 64% (range: 20% ‒ 100%) was shown in one study. Epidemics begin and end suddenly; the duration of disease in a herd is less than 8 days in most cases and not found to exceed 15 days. Some herds, however, suffer from recurrences of disease, suggesting enzootic persistence of the organism (Moxley, 2000).
3.2 Risk factors
The most important risk factor associated to ED is the lack or scarce presence of specific local protective immunity in the small intestines of piglets. The piglets get the typical secretory immunoglobulins (IgA) that prevents E. coli adhering to the small intestine mucosa from the mother’s milk. After weaning, the presence of IgA from the mother’s milk in the piglet gut disappears and then becomes highly susceptible to this disease (Salguero, 2016). For those reasons, ED will normally appear after 1 or 2 weeks post-weaning and most likely associated to the presence of other risk factors as:
- Stress
- Genetic predisposition: Certain breeds have been historically associated to higher incidence of ED. We know that pigs from different genetic backgrounds express different cell receptors associated to the recognition and adherence of specific fimbriae of E. coli. This can be one cause associated to the highly variability in the morbidity of ED in different production scenarios.
- Nutritional factors: Special attention must be taken to percentage of protein in the diet. Piglets can only digest up to 45% of crude protein (value expressed in terms of dry matter). Depending on the production system, the percentage of low digestibility protein in the diet is variable. The higher percentage of low digestibility protein, the higher the risk of gastrointestinal problems.
- Normal gut flora: The difference between piglets of different ages at intestinal level in terms of microbial flora is huge. The animals at weaning will undergo a massive change from liquid to solid diet. Changes in the environment may also induce changes in the intestinal flora easing the proliferation of E. coli (Salguero, 2016).
4. Pathogenicity mechanism
The pathogenicity caused by STEC strains is mediated by genes expressing Shiga toxins (Stx), genes present on the pathogenicity island of the locus of enterocyte effacement (LEE), as well as other genes of interest that encode virulence factors. Another challenge is the hybridization of pathogenic subtypes of other E. coli groups which have acquired the Stx1 from bacteriophages and the Stx2 phage by horizontal gene transfer, such as the O104:H4 serotype involved in foodborne outbreaks (Kyle et al., 2012; Laing et al., 2012).
There are 41 genes located on the pathogenicity island of the LEE, divided into 5 operons, named LEE-1 to LEE-5. Regions 1, 2, and 3 encode the type III secretory system (T3SS) that translocates bacterial protein effectors into the enterocyte. LEE-4 contains the EspADB gene encoding translocating proteins, forming a channel through which T3SS is able to provide proteins to host cells (attaching and effacing). The adhesion genes are located in LEE-5, including translocated intimin receptor (TIR), EaeA for Intimin, and CesT for the TIR chaperone (Elliott et al., 2000; Lara-Ochoa et al., 2010).
Bacterial attachment, through the proteins produced by the LEE, causes deep histological changes, due to the destruction of the microvilli of the intestinal epithelium, modifying the morphology of the apical zone of enterocytes (Vidal and others 2007). After initial contact, the bacterium injects a set of proteins with cytotoxic action into the cell, which bind through the T3SS, which acts as a bridge between the microorganism and the cell. Cellular infection by STEC is completed with the binding of Tir to Intimin, a protein encoded by the eaeA gene, which causes an intimate and resistant bond between the bacterium and the cell through a fixation pedestal formed by the accumulation of actin, the result of alterations in the cellular cytoskeleton (Lara-Ochoa et al., 2010).
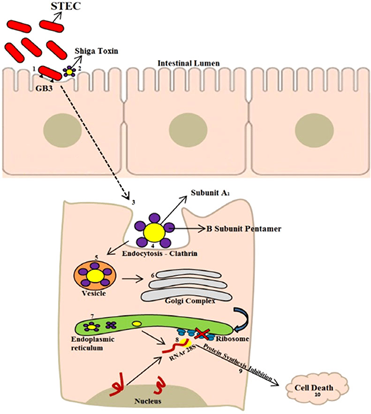
Negative LEE serotypes may display a binding of the autoagglutinating adhesin encoded by the Saa gene, which enables the binding of the bacteria to the cell. This adhesin was first described in a LEE-negative strain (O113:H21), responsible for an outbreak of hemolytic uremic syndrome (HUS). After binding, STEC strains can initiate Shiga toxin-production, being able to express 1, both, or variants such as stx1 (stx1a, stx1c, and stx1d) and stx2 (stx2a, stx2b, stx2c, stx2d, stx2e, stx2f, and stx2g). Stx2 is the main toxin involved in the most severe cases of the human disease. The Shiga toxin is formed by a subunit A, which exerts toxic action and inhibits the protein synthesis of cells, and by another 5 subunits B, which are responsible for the binding of the toxin to the host cell globotriacylceramide (Gb3) receptor. Portion A of the toxin is internalized in the cell and is transported to the Golgi complex and endoplasmic reticulum. This subunit is N-glycosidase, which will act on the 28s portion of messenger RNA and also on the ribosomal subunit, inhibiting protein synthesis and causing cell apoptosis (Figure 3). Endothelial damage compromises physiological functions including renal function, with cell swelling and separation from the basal membrane, fibrin, and thrombi, resulting in the narrowing of the capillary lumen, consequently reducing the blood supply to the glomeruli leading to impaired renal function.
Usually, the symptoms appear after a short incubation period, of 3 to 4 days, where the patient initially develops diarrhea, accompanied by abdominal pain, which, in most cases, aggravates to bloody diarrhea. The use of antibiotics is not indicated, due to the increased risk of HUS. In addition, the use of growth-promoting substances, such as antibiotics, may contribute to the dissemination of pathogenicity genes and to the emergence of new pathogenic serotypes (Köhler et al., 2000).
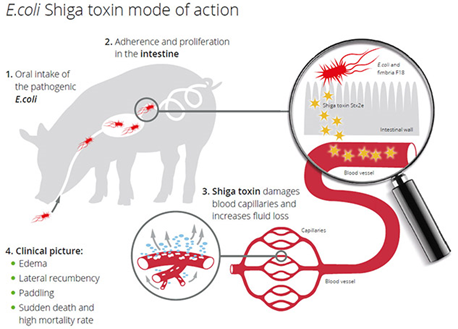
The causative agent of ED is Shigatoxin-2e. It is encoded and produced by a specific virotype of E. coli and occurs worldwide in diseased pig herds as well as in healthy herds. The primary habitat of E. coli in pigs is the gastrointestinal tract. Outside the intestine E. coli are found in fecal-contaminated feed or water, water from water pipes contaminated by biofilm, soil, and the environment of the pig barn. The infection happens via the oral route (Figure 4). When ingested in sufficient numbers, E. coli causing ED colonize and then proliferate rapidly to attain massive numbers. Colonization develops over 3-6 days and requires attachment of fimbrial adhesins to complementary receptors on the small intestinal epithelium or in the mucus coating in the distal jejunum and ileum. Stx2e is absorbed into the circulation and causes vascular damage in target organs.
The degenerative angiopathy of small arteries and arterioles results in an increase in vascular permeability. Due to osmosis, fluid leaves the vascular system and diffundates into various tissues, causing lesions that can be seen macroscopically as edema. Gelatinous edema appears in the submucosa of the gastric cardia and occasionally in the fundus, in the mesocolon, in the small intestinal mesentery and gallbladder bed. The mesenteric and colic lymph nodes may be swollen, edematous and congested. Edematous swelling of eyelids, forehead, larynx and brain can be observed (Zimmerman et al., 2012)
5. Clinical signs and pathology
5.1 Clinical signs
Clinically, acute neurological signs consisting of depression, staggering gait and tremor represent the hallmarks of ED. Due to the oedema in the brain pigs show convulsions, ataxia and lateral recumbency with paddling of limbs (Figure 5). Few pigs survive the acute disease, but those that do remain runts (i.e. show stunted growth).


These are followed by lateral recumbence, with characteristic rhythmic limb paddling, dyspnea, extensor muscle rigidity, convulsions and finally flaccid paralysis, coma and death, in most cases within 24 h following the appearance of the clinical signs (MacLeod et al., 1991; Zimmerman et al., 2012). After clinical debut, the mortality is generally high (up to 90%), being influenced by both STEC strain pathogenicity and individual susceptibility. This neurological signs are associated with various degrees of eyelid, face (mainly forehead) and laryngeal edema, although these clinical signs can be easily missed due to the mild severity of most cases and the peculiarities of swine husbandry systems. Subclinical cases of ED, in which the characteristic vascular lesions are histological present, but without the above described clinical signs, are reported. Diarrhea and hyperthermia may be present in some ED episodes, enterotoxins being also produced by some STEC. Constipation can also occasionally be observed in ED affected pigs (Zimmerman et al., 2012).
5.2 Pathology
Postmortem findings
During postmortem examination the typical diffuse, vasculogenic edema, occasionally accompanied with petechia (due to microthrombosis that follows the vascular endothelial disruption) can be present in most tissues. Most often, the edema affects the digestive system (gastric lamina propria and submucosa, spiral colon wall, meso-colon, small intestine mesentery and mesenteric lymph nodes and gallbladder) (Figure 7), skin and subcutis (palpebrae, frontal skin, submandibular, ventral abdomen) and adjacent lymph nodes, thoraco-abdominal and pericardial serosa consisting of moderate to abundant effusions, often presented as protein rich/modified transudates (Tabaran and Tabaran, 2019).

A – section of the large gastric curve; severe edema of the gastric wall (indicated by arrows), presented as a gelatinous material diffusely expanding the submucosa. B and C – diffuse “gelatinous” edema of the mesocolon (arrows), associated with segmental colonic-serosa reaction. D – pleural effusion (arrow) (hydrothorax) with secondary, mild pulmonary collapse. E – pericardial effusion rich in protein clots (arrow). F – diffuse, bilateral, mild, pulmonary congestion and edema (asterisk) accompanied with mediastinal edema G – the forehead subcutis is locally expanded by edema (asterisk), mainly in areas adjacent to the eyelids. H – eye and adnexa: edema of the eyelids and mild congestion of the bulbar conjunctiva. I – diffuse, minimal, cerebral congestion and edema.
Hemorrhagic gastroenteritis is also occasionally observed. The respiratory system (larynx, and lungs) and lower urinary tract are occasionally affected. The cerebrum is responsible for most of the dramatic and diagnostically suggestive clinical signs (cerebrospinal angiopathy/swine cerebral angiopathy) which accompany edema disease. However, macroscopically changes are often absent or minimal to mild and consist of meningeal congestion and petechias, brain edema (wide gyri and diffusely shallowed sulci) and occasionally symmetric neural cerebral malacia (focal symmetric encephalomalacia). The neuroanatomical distribution of the malacic lesions in ED are medulla oblongata, diencephalon (mainly thalamus), and basal nuclei (i.e. caudate nucleus, putamen, substantia nigra, globus pallidus, subthalamic nucleus, etc.). Mild cerebellar edema and hemorrhage are frequent findings in experimental ED (MacLeod et al., 1991). Gastric ulceration of the esophageal (cardia) region is occasionally present in animals which survive the acute phase of ED (Clugston et al., 1974).
An interesting change is represented by the bilateral, acute renal cortical necrosis. However, it presents low specificity for ED since it was not experimentally reproduced by MacLeod et al (1991), following intravenous administration of purified Stx2e. This is due to a Shwartzman like reaction mimicking the massive renal glomerular damage observed in humans in the hemolytic uremic syndrome (HUS), induced by O157:H7, enterohemorrhagic E. coli.
All the above described changes are due to the systemic action of the Stx2e and reflect the distribution of the cellular membrane receptors for this toxin. The presence and distribution of these receptors dictate the pathological features (anatomical distribution of the arteriolar hyaline degeneration) and clinical signs and outcome of edema disease cases (Tabaran and Tabaran, 2019).
Histopathologic findings
Histologically, the key lesions of ED are considered to be directly induced by Stx2e and consist of segmental (occasionally transmural) fibrinoid necrosis affecting the small arteries and arteriolar media (smooth muscle cells) (“arteriolar hyaline degeneration”), with endothelial disruption, and intraluminal mural (occluding or sub-occluding) fibrinous thrombi. The lesions described above are associated with secondary changes induced by vascular disruption (as vasculogenic, severe edema, congestion and hemorrhage), ischemia and infarction (especially important within the central nervous system). A leukocytic infiltrate (lympho-histiocytic and neutrophilic) can also be occasionally admixed with the fibrin and cell debris within the necrotic arteriolar walls. Ocular lesions consisting of retinal edema and hemorrhages are reported by MacLeod et al (1991), in experimental cases of ED. Colonic and cecal microerosions with no associated inflammation are occasionally reported, being most likely the consequence of enteral focal ischemia secondary to endovascular microthrombosis. Ultrastructurally, within the affected areas, endothelial cells are swelled and vacuolated, with subendothelial deposition of fibrin (electron dense material), discontinuous plasma membranes, with cytoplasmic condensation, reduced number of mitochondria and decreased endoplasmic reticulum, chromatin clumping and margination. Endothelial proliferation is rarely observed and present as early as 3 days following STEC (O139: K12: H1) inoculation in gnotobiotic piglets. The arteriolar necrosis can be occasionally observed in the medial myocytes without significant disruption of the vascular endothelium. Within the small intestine, the Stx2e-producing E. coli are occasionally present as rod shaped Gram negative (“coliform”) bacteria adherent on the apical domain of the enterocytes (Tabaran and Tabaran, 2019).
6. Diagnosis
Differential diagnoses of the disease in relation to central nervous system disorders include: pseudorabies; teschoviral encephalitis; Streptococcus suis or Haemophilus parasuis induced meningitis among others. Farms with suspected S. suis infections in nursery unit which are not responding well on treatment of choice, such as amoxicillin, should be evaluated for the presence of STEC. It is important to note that with ED infection there will be no meningitis at histology. There are several non-infectious sources that may present similar clinical signs, such as water deprivation (salt poisoning), vitamin E/selenium or even stress due to handling. The disease should also be included as a differential diagnosis when sudden death is observed in the first weeks after the weaning (Zimmerman et al., 2012). For these reasons, diagnosis is an important tool to identify the main cause of disease and the conclusion should not be reached based only on clinical signs and farm history. When completing farm sampling, the number of samples required depends on the expected prevalence of the disease and the total number of animals at each age on farm. Faecal samples and swabs can be taken for culture, but no more than five individual faecal samples should be pooled together. In order to confirm STEC circulation on farm usually six pools per five piglets from six different pens in the nursery is recommended. In deceased pigs, the collection of jejunum parts or swabs from the mucosa of the jejunum is highly recommended.
The first step with these samples is to perform the analysis of the bacterial culture. Once the E. coli strain is cultured, multiplex PCR can be carried out for the detection of fimbrial and toxin genes. The advantage of the multiplex PCR is to deliver a result on the spectrum of adhesion factors and toxins which enable the veterinarian to detect the E. coli strains involved and to make the correct diagnosis. It is necessary to provide the laboratory with a complete clinical history, including the clinical signs observed in the affected animals and the presumed aetiological cause (Gale and Velazquez, 2020).
During the post-mortem examination, the typical diffuse, vasculogenic oedema, occasionally accompanied with petechia, can be present in most tissues. Most often, the oedema affects the digestive system (gastric lamina propria and submucosa, spiral colon wall, meso-colon, small intestine mesentery and mesenteric lymph nodes and gallbladder), skin and subcutis (palpebrae, frontal skin, submandibular, ventral abdomen) and adjacent lymph nodes, thoraco-abdominal and pericardial serosa. Histopathological study of the samples, particularly the brain and distal jejunum, would help to clarify the diagnosis if the lesions of the general pathology are unspecific, and also to assess the relevance of the results obtained by culture and PCR. By microscopic examination, lesions of oedema, haemorrhage and vasculitis are noted. Microscopic lesions are associated with vascular injury and include vessel necrosis, perivascular oedema and haemorrhage, and superficial colonic and caeca erosions. The vascular lesions are observed in the cerebellar folia, submucosa and mucosa of the stomach, caecum, colon, and sporadically in the retina (MacLeod et al., 1991).
7. Vaccination
The Shiga toxin vaccination reduces the mortality and clinical signs of ED into the finishing period. The single-dose of vaccination can be used from 4 days of age. Three weeks after the vaccination animals are protected by neutralising antibodies, which persist until at least the 15th week of life and protect animals in the most vulnerable period of life. Nothing is as natural as the pig’s own immunity, but the inherent immunity against Shiga toxin can be actively boosted by vaccination. These antibodies neutralise the Shiga toxin and therefore prevent the development of acute or chronic clinical signs. However, the presence and colonisation of animals by Shiga toxin producing E. coli in the herd is not reduced by the vaccine because it does not act against the bacterium itself. While the vaccination itself almost eliminates the rate of losses and runts resulting from ED, additionally the farmer can dramatically reduce the use of antimicrobials or ZnO for controlling STEC in pigs. The Shiga toxin vaccination helps to minimise the risk of developing resistant bacteria and thus actively contributes to consumer protection. Compliance and efficiency of vaccination is easy to control through use of an in-house serum neutralisation test. The result of such a test corresponds directly to protection of piglets after vaccination. In Europe, a vaccine based on a recombinant and genetically modified toxoid has been developed, which consists of a subunit in which the antigen is the genetically modified Stx2e recombinant toxin (Gale and Velazquez, 2020).
8. Control and prevention
Control and prevention of ED can be difficult, as actually, few commercial vaccines or effective treatments are available. Nutritional changes can be made which can help to reduce colonisation with E. coli in pigs after weaning. For example, supplementation of piglets’ diet with ZnO can reduce post-weaning diseases through reducing E. coli adhesion and intestinal permeability. The use of high levels (pharmacological level) of ZnO post-weaning will be banned in countries in EU mid-2022, thus other strategies will be needed. Feeding of high energy and protein content that is administered after weaning causes an increase in the pH of the intestine that favours the multiplication of E. coli. Therefore, limiting the amount of feed or ad libitum feeding of high fibre and low protein diet can modulate fimbrial receptors (Kelly et al., 1994), which can reduce colonisation by E. coli after weaning.
However, these strategies usually limit growth potential of modern pig genetics. Antibiotics have historically been used to control outbreaks, however due to the rapid course of the disease, treatment is often too late for piglets with clinical signs. The most effective antibiotics and classes of antibiotics frequently used postweaning, such as colistin or fluorochinolones, belong according to new European classification to category B (‘restrict’). Antibiotics in this category are critically important in human medicine and their use in animals should be restricted to mitigate the risk to public health. The best results and optimal ED prevention and reduction of mortality have been achieved with vaccination, with vaccines containing the modified, and therefore non-toxic, Shiga toxin (Casanova et al., 2018).
9. References
Bélanger, L., Garenaux, A., Harel, J., Boulianne, M., Nadeau, E., Dozois, C.M., 2011. Escherichia coli from animal reservoirs as a potential source of human extraintestinal pathogenic E. coli. FEMS Immunology & Medical Microbiology 62, 1-10.
Casanova, N.A., Redondo, L.M., Dailoff, G.C., Arenas, D., Miyakawa, M.E.F., 2018. Overview of the role of Shiga toxins in porcine edema disease pathogenesis. Toxicon 148, 149-154.
Castro, V.S., Carvalho, R.C.T., Conte‐Junior, C.A., Figuiredo, E.E.S., 2017. Shiga‐toxin producing Escherichia coli: pathogenicity, supershedding, diagnostic methods, occurrence, and foodborne outbreaks. Comprehensive Reviews in Food Science and Food Safety 16, 1269-1280.
Clugston, R.E., Nielsen, N., Smith, D., 1974. Experimental edema disease of swine (E. coli enterotoxemia) III. Pathology and pathogenesis. Canadian Journal of Comparative Medicine 38, 34.
Coddens, A., Verdonck, F., Tiels, P., Rasschaert, K., Goddeeris, B., Cox, E., 2007. The age-dependent expression of the F18+ E. coli receptor on porcine gut epithelial cells is positively correlated with the presence of histo-blood group antigens. Veterinary microbiology 122, 332-341.
Elliott, S.J., Sperandio, V., Girón, J.A., Shin, S., Mellies, J.L., Wainwright, L., Hutcheson, S.W., McDaniel, T.K., Kaper, J.B., 2000. The locus of enterocyte effacement (LEE)-encoded regulator controls expression of both LEE-and non-LEE-encoded virulence factors in enteropathogenic and enterohemorrhagic Escherichia coli. Infection and immunity 68, 6115-6126.
Fricke, R., Bastert, O., Gotter, V., Brons, N., Kamp, J., Selbitz, H.-J., 2015. Implementation of a vaccine against Shigatoxin 2e in a piglet producing farm with problems of Oedema disease: case study. Porcine Health Management 1, 1-5.
Gale, C., Velazquez, E., 2020. Oedema disease: a review of the disease and control and preventative measures. Livestock 25, 142-147.
Helgerson, A.F., Sharma, V., Dow, A.M., Schroeder, R., Post, K., Cornick, N.A., 2006. Edema disease caused by a clone of Escherichia coli O147. J Clin Microbiol 44, 3074-3077.
Kaper, J., Nataro, J., Mobley, H., 2004. Pathogenic Escherichia coli Nature Reviews Microbiology. London.
Kelly, D., Begbie, R., King, T., 1994. Nutritional influences on interactions between bacteria and the small intestinal mucosa. Nutrition Research Reviews 7, 233-257.
Köhler, B., Karch, H., Schmidt, H., 2000. Antibacterials that are used as growth promoters in animal husbandry can affect the release of Shiga-toxin-2-converting bacteriophages and Shiga toxin 2 from Escherichia coli strains. Microbiology 146, 1085-1090.
Kyle, J.L., Cummings, C.A., Parker, C.T., Quiñones, B., Vatta, P., Newton, E., Huynh, S., Swimley, M., Degoricija, L., Barker, M., 2012. Escherichia coli serotype O55: H7 diversity supports parallel acquisition of bacteriophage at Shiga toxin phage insertion sites during evolution of the O157: H7 lineage. Journal of bacteriology 194, 1885-1896.
Laing, C.R., Zhang, Y., Gilmour, M.W., Allen, V., Johnson, R., Thomas, J.E., Gannon, V.P., 2012. A comparison of Shiga-toxin 2 bacteriophage from classical enterohemorrhagic Escherichia coli serotypes and the German E. coli O104: H4 outbreak strain. PLoS One 7, e37362.
Lara-Ochoa, C., Oropeza, R., Huerta-Saquero, A., 2010. Regulation of the LEE-pathogenicity island in attaching and effacing bacteria. Current research, technology and education topics in applied microbiology and microbial biotechnology 1, 635-645.
Lillie-Jaschniski, K., Hillen, S., Lindner, T., 2016. Losses and amount of antimicrobial treatment due to oedema disease effect of vaccination with Ecoporc SHIGA evaluated on 179 German farms. ESPHM/IVPS, Dublin, 7-10.
MacLeod, D., Gyles, C., Wilcock, B., 1991. Reproduction of edema disease of swine with purified Shiga-like toxin-II variant. Veterinary Pathology 28, 66-73.
Moxley, R.A., 2000. Edema disease. Veterinary Clinics of North America: Food Animal Practice 16, 175-185.
Paton, J.C., Paton, A.W., 1998. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clinical microbiology reviews 11, 450-479.
Reeves, P.R., Hobbs, M., Valvano, M.A., Skurnik, M., Whitfield, C., Coplin, D., Kido, N., Klena, J., Maskell, D., Raetz, C.R., 1996. Bacterial polysaccharide synthesis and gene nomenclature. Trends in microbiology 4, 495-503.
Salguero, J., 2016. Risk factors associated to Oedema disease in pigs. Professional Pig Community.
Tabaran, F., Tabaran, A., 2019. Edema disease of swine: A review of the pathogenesis. Porcine Research 9, 7-14.
Wang, L., Briggs, C.E., Rothemund, D., Fratamico, P., Luchansky, J.B., Reeves, P.R., 2001. Sequence of the E. coli O104 antigen gene cluster and identification of O104 specific genes. Gene 270, 231-236.
Zimmerman, J.J., Karriker, L.A., Ramirez, A., Stevenson, G.W., Schwartz, K.J., 2012. Diseases of swine. John Wiley & Sons.